Finding the ground state of a dual-species Bose-Einstein condensate#
This example demonstrates the calculation of the ground state of a dual-species Bose-Einstein condensate. We use the Gross-Pitaevskii equation (GPE) to describe the dynamics of the two wavefunctions and perform an imaginary time evolution using the split-step method to find the ground state. For further details and for the specific configuration used in this example, refer to: Pichery et al., AVS Quantum Sci. 5, 044401 (2023); doi: 10.1116/5.0163850.
Since this example uses three dimensions it requires high performance and we recommend running it on a GPU in the examplescuda environment. For this reason it is also written in JAX with jax.lax.scan.
Imports and setup#
Import basic libraries and enable float64 precision types in JAX.
[1]:
from functools import reduce
from typing import (
Dict, Iterable, Any, Optional, Tuple, TypedDict
)
import os
from scipy import constants # type: ignore
import numpy as np
import jax.numpy as jnp
from jax import config
import fftarray as fa
# Enable double float precision for jax
config.update("jax_enable_x64", True)
Physical setup#
Define the potential between the two interacting atomic species and basic physical constants.
[2]:
# --------------------
# Physical constants
# --------------------
hbar: float = constants.hbar
a_0: float = constants.physical_constants['Bohr radius'][0]
kb: float = constants.Boltzmann
# coupling constant (used in GPE)
def coupling_fun(m_red: float, a: float) -> float:
return 2 * np.pi * hbar**2 * a / m_red
# Rubidium 87
m_rb87: float = 86.909 * constants.atomic_mass # The atom's mass in kg.
a_rb87: float = 98 * a_0 # s-wave scattering length
# coupling constant (used in GPE)
coupling_rb87: float = coupling_fun(0.5*m_rb87, a_rb87)
# Potassium 41
m_k41: float = 40.962 * constants.atomic_mass # The atom's mass in kg.
a_k41: float = 60 * a_0 # s-wave scattering length
# coupling constant (used in GPE)
coupling_k41: float = coupling_fun(0.5*m_k41, a_k41)
# Interspecies interaction
a_rb87_k41: float = 165.3 * a_0
reduced_mass_rb87_k41 = m_rb87 * m_k41 / (m_rb87 + m_k41)
# coupling constant (used in GPE)
coupling_rb87_k41: float = coupling_fun(reduced_mass_rb87_k41, a_rb87_k41)
# Define dual species GPE potentials
def gpe_potential_two_species(
psi_pos_sq_1: fa.Array,
psi_pos_sq_2: fa.Array,
coupling_constant_1: float,
coupling_constant_12: float,
trap_potential_1: fa.Array,
num_atoms_1: float,
num_atoms_2: float,
) -> fa.Array:
"""
Calculate the 2-species GPE potential for species number 1.
This does not include the energies that only depend on the other species.
This does not include the kinetic energy.
"""
self_interaction = num_atoms_1 * coupling_constant_1 * psi_pos_sq_1
interaction_12 = num_atoms_2 * coupling_constant_12 * psi_pos_sq_2
return self_interaction + interaction_12 + trap_potential_1
Quantum mechanics helpers#
These functionalities can also be found in the matterwave library.
Get ground state of quantum harmonic oscillator.#
This function creates an fa.Array from the given coordinates and trap frequencies.
[3]:
def ground_state_ho(
mass: float,
omegas: Iterable[float],
pos_coords: Iterable[fa.Array],
) -> fa.Array:
psi = fa.full([], "pos", 1.0, xp=jnp)
for omega, pos_1d in zip(omegas, pos_coords, strict=True):
psi = (
psi * (omega / (np.pi*hbar))**(1/4)
* fa.exp(-mass * omega * (pos_1d**2)/(2*hbar))
)
norm = fa.integrate(fa.abs(psi)**2)
return psi / fa.sqrt(norm)
Imaginary time propagation#
A basic implementation of imaginary time propagation using the split-step method.
[4]:
def split_step_imaginary_time(
psi: fa.Array,
V: fa.Array,
dt: float,
mass: float,
) -> fa.Array:
"""Perform an imaginary time split-step of second order in VPV configuration."""
# Calculate half step imaginary time potential propagator
V_prop = fa.exp((-0.5*dt / hbar) * V)
# Calculate full step imaginary time kinetic propagator (k_sq = kx^2 + ky^2 + kz^2)
k_sq = reduce(lambda a,b: a+b, [
(2*np.pi * fa.coords_from_dim(dim, "freq", xp=jnp, dtype=jnp.float64))**2
for dim in psi.dims
])
T_prop = fa.exp(-dt * hbar * k_sq / (2*mass))
# Apply half potential propagator
psi = V_prop * psi.into_space("pos")
# Apply full kinetic propagator
psi = T_prop * psi.into_space("freq")
# Apply half potential propagator
psi = V_prop * psi.into_space("pos")
# Normalize after step
state_norm = fa.integrate(fa.abs(psi)**2)
psi = psi / fa.sqrt(state_norm)
return psi
Energy computation#
Compute the kinetic and potential energy of a given wave function.
[5]:
def get_e_kin(
psi: fa.Array,
mass: float,
):
"""Calculate the kinetic energy of the wavefunction."""
# Calculate k^2 = (2πf)^2
ksq = reduce(lambda a,b: a+b, [
(2*np.pi * fa.coords_from_dim(dim, "freq", xp=jnp, dtype=jnp.float64))**2
for dim in psi.dims
])
post_factor = hbar**2 / (2*mass)
# Calculate |ψ(f)|^2
wf_abs_sq = fa.abs(psi.into_space("freq"))**2
# Calculate E_kin = <ψ|(hbar*k)^2/2m|ψ> = ∫|ψ(f)|^2 * k^2 df * hbar^2 / (2m)
return fa.integrate(wf_abs_sq * ksq).values("freq") * post_factor
def get_e_pot(
psi: fa.Array,
V: fa.Array,
):
return fa.integrate(
fa.abs(psi.into_space("pos"))**2 * V,
dtype="float64"
).values("pos") / (kb * 1e-6)
Definition of the state initialization and the step function#
The simulation loop is in this example written with jax.lax.scan in order to achieve high performance, especially on a GPU.
calc_ground_state_two_species sets up the domain and the physical problem and then runs imaginary_time_step_dual_species in a loop to execute the optimization.
[6]:
from jax.lax import scan
# Register fftarray pytree nodes for JAX
try:
fa.jax_register_pytree_nodes()
except ValueError:
print(
"JAX pytree nodes registration failed. " \
"Probably due to being already registered."
)
class DualSpeciesProperties(TypedDict):
psi_rb87: fa.Array
psi_k41: fa.Array
rb_potential: fa.Array
k_potential: fa.Array
num_atoms_rb87: float
num_atoms_k41: float
def calc_ground_state_two_species(
N_iter: int,
plot_dir: Optional[str] = None,
) -> Tuple[DualSpeciesProperties, Dict[str, Any]]:
if plot_dir and not os.path.exists(plot_dir):
os.makedirs(plot_dir)
dt_list = np.full(N_iter, 5e-7)
# Number of atoms
num_atoms_rb87 = 43900
num_atoms_k41 = 14400
rb_omega_x = 2*np.pi * 24.8 # rad/s
rb_omega_y = 2*np.pi * 378.3 # rad/s
rb_omega_z = 2*np.pi * 384.0 # rad/s
k_omega_x = rb_omega_x * np.sqrt(m_rb87/m_k41)
k_omega_y = rb_omega_y * np.sqrt(m_rb87/m_k41)
k_omega_z = rb_omega_z * np.sqrt(m_rb87/m_k41)
# --------------------
# fftarray definitions
# --------------------
# Define dimensions
x_dim = fa.dim_from_constraints(
"x",
pos_extent=400e-6,
n=2**9,
freq_middle=0.,
pos_middle=0.,
dynamically_traced_coords=False,
)
y_dim = fa.dim_from_constraints(
"y",
pos_extent=50e-6,
n=2**8,
freq_middle=0.,
pos_middle=0.,
dynamically_traced_coords=False,
)
z_dim = fa.dim_from_constraints(
"z",
pos_extent=50e-6,
n=2**7,
freq_middle=0.,
pos_middle=0.,
dynamically_traced_coords=False,
)
# Define 1d arrays
x: fa.Array = fa.coords_from_dim(x_dim, "pos", xp=jnp, dtype=jnp.float64)
y: fa.Array = fa.coords_from_dim(y_dim, "pos", xp=jnp, dtype=jnp.float64)
z: fa.Array = fa.coords_from_dim(z_dim, "pos", xp=jnp, dtype=jnp.float64)
# Define 3d arrays for potential
rb_potential = 0.5 * m_rb87 * (
rb_omega_x**2 * x**2
+ rb_omega_y**2 * y**2
+ rb_omega_z**2 * z**2
)
k_potential = 0.5 * m_k41 * (
k_omega_x**2 * x**2
+ k_omega_y**2 * y**2
+ k_omega_z**2 * z**2
)
# Define 3d arrays for initial wavefunction
init_psi = fa.full((x_dim, y_dim, z_dim), "pos", 1, xp=jnp, dtype=jnp.float64)
state_norm = fa.integrate(fa.abs(init_psi)**2)
init_psi_rb = init_psi / fa.sqrt(state_norm)
init_psi_k = init_psi / fa.sqrt(state_norm)
# When using jax.lax.scan, the input fa.Array must have the same properties as
# the output one. As the scanned method imaginary_time_step_dual_species
# returns the fa.Array with space="pos" and factors_applied=False,
# we transform the input state accordingly.
init_psi_rb = init_psi_rb.into_space("pos").into_factors_applied(False)
init_psi_k = init_psi_k.into_space("pos").into_factors_applied(False)
init_properties: DualSpeciesProperties = {
"psi_rb87": init_psi_rb,
"psi_k41": init_psi_k,
"rb_potential": rb_potential,
"k_potential": k_potential,
"num_atoms_rb87": num_atoms_rb87,
"num_atoms_k41": num_atoms_k41
}
res: Tuple[DualSpeciesProperties, Dict[str, Any]] = scan(
f=imaginary_time_step_dual_species, # type: ignore
init=init_properties,
xs=dt_list,
)
return res
def imaginary_time_step_dual_species(
properties: DualSpeciesProperties,
dt: float,
) -> Tuple[DualSpeciesProperties, Dict[str, float]]:
"""
Perform a single imaginary time step for the dual species GPE.
Additionally, calculate all relevant energies.
The states are returned in position space.
"""
psi_rb87 = properties["psi_rb87"]
psi_k41 = properties["psi_k41"]
rb_potential = properties["rb_potential"]
k_potential = properties["k_potential"]
num_atoms_rb87 = properties["num_atoms_rb87"]
num_atoms_k41 = properties["num_atoms_k41"]
## Calculate the potential energy operators (used for split-step and plots)
psi_rb87 = psi_rb87.into_space("pos")
psi_k41 = psi_k41.into_space("pos")
psi_pos_sq_rb87 = fa.abs(psi_rb87)**2
psi_pos_sq_k41 = fa.abs(psi_k41)**2
self_interaction_rb87 = num_atoms_rb87 * coupling_rb87 * psi_pos_sq_rb87
interaction_rb87_k41 = num_atoms_k41 * coupling_rb87_k41 * psi_pos_sq_k41
V_rb87 = self_interaction_rb87 + interaction_rb87_k41 + rb_potential
self_interaction_k41 = num_atoms_k41 * coupling_k41 * psi_pos_sq_k41
interaction_k41_rb87 = num_atoms_rb87 * coupling_rb87_k41 * psi_pos_sq_rb87
V_k41 = self_interaction_k41 + interaction_k41_rb87 + k_potential
## Calculate energies for plotting and convergence check
# Calculate potential energy (in µK)
E_pot_rb87 = get_e_pot(psi_rb87, V_rb87)
E_pot_k41 = get_e_pot(psi_k41, V_k41)
# Calculate kinetic energy (in µK)
E_kin_rb87: float = get_e_kin(psi_rb87, mass=m_rb87) / (kb * 1e-6)
E_kin_k41: float = get_e_kin(psi_k41, mass=m_k41) / (kb * 1e-6)
# Calculate the total energy
E_tot = E_kin_rb87 + E_kin_k41 + E_pot_rb87 + E_pot_k41
## Imaginary time split step application
psi_rb87 = split_step_imaginary_time(
psi=psi_rb87,
V=V_rb87,
dt=dt,
mass=m_rb87,
)
psi_k41 = split_step_imaginary_time(
psi=psi_k41,
V=V_k41,
dt=dt,
mass=m_k41,
)
return properties | {"psi_rb87": psi_rb87, "psi_k41": psi_k41}, {
"E_kin_rb87": E_kin_rb87,
"E_kin_k41": E_kin_k41,
"E_pot_rb87": E_pot_rb87,
"E_pot_k41": E_pot_k41,
"E_tot": E_tot
}
Run the simulation#
This cell takes about 1 minute for the 5000 steps on an NVIDIA A100 GPU and may take a long time when only running on a CPU.
[ ]:
# Reducing the number of steps in test mode
# allows running this notebook as part of the test suite.
if os.environ.get("TEST_MODE", "False") == "True":
N_iter=2
else:
# This branch is taken when executed in a normal notebook
N_iter=5000
final_properties, energies = calc_ground_state_two_species(N_iter=N_iter)
Plotting the results#
[8]:
import matplotlib.pyplot as plt
from bokeh.plotting import show
from bokeh.io import output_notebook
from helpers import plt_integrated_1d_densities, plt_integrated_2d_density
output_notebook(hide_banner=True)
Final state#
We normalize the final states to their respective number of atoms:
[9]:
rb_ground_state = (
final_properties["psi_rb87"].into_space("pos")
* np.sqrt(final_properties["num_atoms_rb87"])
)
k_ground_state = (
final_properties["psi_k41"].into_space("pos")
* np.sqrt(final_properties["num_atoms_k41"])
)
Integrating in the \(z\) dimension yields a two dimensional plot per species and space. Use the zoom tool in the plots to zoom in on the details of the simulation.
[10]:
show(
plt_integrated_2d_density(
rb_ground_state,
red_dim_name="z",
data_name="Rb87",
title_prefix="N",
)
)
[11]:
show(
plt_integrated_2d_density(
k_ground_state,
red_dim_name="z",
data_name="K41",
title_prefix="N",
)
)
Integrating in \(y\) and \(z\) yields allows us to plot the probability densities of both species:
[12]:
show(
plt_integrated_1d_densities(
arrs={"Rb87": rb_ground_state, "K41": k_ground_state},
red_dim_names=["y", "z"],
# Zoom in a bit to see the spatial structures better.
x_range_pos=(-4e-5, +4e-5),
x_range_freq=(-1e5, +1e5),
y_label_prefix="N",
)
)
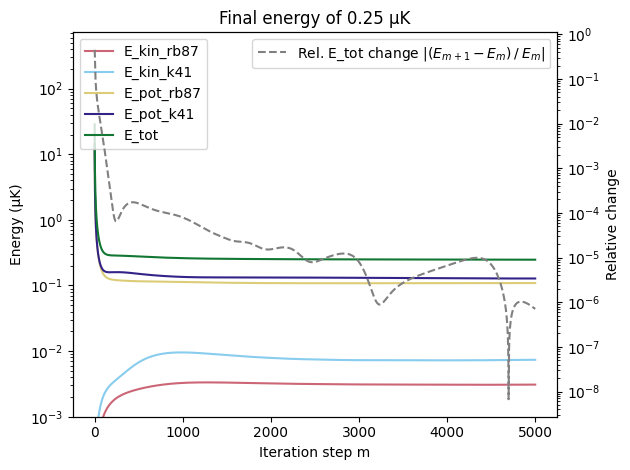
Energies as a function of iteration steps#
[13]:
COLORS = ["#CC6677", "#88CCEE", "#DDCC77", "#332288", "#117733"]
fig, ax1 = plt.subplots()
ax1.plot(energies["E_kin_rb87"], label="E_kin_rb87", color=COLORS[0])
ax1.plot(energies["E_kin_k41"], label="E_kin_k41", color=COLORS[1])
ax1.plot(energies["E_pot_rb87"], label="E_pot_rb87", color=COLORS[2])
ax1.plot(energies["E_pot_k41"], label="E_pot_k41", color=COLORS[3])
ax1.plot(energies["E_tot"], label="E_tot", color=COLORS[4])
ax1.set_title(f"Final energy of {energies['E_tot'][-1]:.2f} µK")
ax1.set_xlabel("Iteration step m")
ax1.set_ylabel("Energy (µK)")
ax1.set_yscale("log")
ax1.set_ylim(bottom=1e-3)
ax1.legend(loc="upper left")
ax2 = ax1.twinx()
relative_change = np.abs(np.diff(energies["E_tot"]) / energies["E_tot"][:-1])
ax2.plot(
np.arange(1, len(energies["E_tot"])),
relative_change,
color="gray",
linestyle="--",
label=r"Rel. E_tot change $|(E_{m+1}-E_m) \:/ \: E_{m}|$",
)
ax2.set_ylabel("Relative change")
ax2.set_yscale("log")
ax2.legend(loc="upper right")
plt.tight_layout()